Laboratory of Norbert Perrimon, Ph.D.

Communication between cells, tissues and organs in Drosophila
Using Drosophila as a model, the Perrimon lab investigates intercellular and inter-organ communication. To support this, the lab develops tools in functional genomics and proteomics, including the Gal4-UAS system, FLP-FRT germline clone method, genome-wide RNAi and CRISPR platforms, and in vivo proximity labeling. Major research contributions include advances in signaling pathways, cell polarity, gut regeneration, and inter-organ signaling.
Interorgan Communication
Our lab investigates organ-to-organ communication using Drosophila as a model, leveraging conserved physiology and powerful genetic tools, including tissue-specific RNAi. Through genetic screens, we’ve identified key secreted factors—such as ImpL2, Upd2, and Pvf1—that mediate systemic signaling. We complement this with transcriptomic (RNAseq, snRNAseq) and proteomic approaches to define how organs respond to stress, diet, and aging. Additionally, we developed a novel TurboID-based proximity labeling method to map secreted proteins and their targets. Together, our work aims to uncover how dysfunction in one tissue influences others, informing mechanisms underlying complex diseases like diabetes, aging, and cancer.

Organ Wasting/Cachexia
Our lab uses Drosophila to model tumor-induced organ wasting relevant to cancer cachexia. We induce gut tumors by overexpressing yorkie/YAP in intestinal stem cells, leading to systemic wasting via tumor-secreted ImpL2, which reduces insulin signaling and disrupts energy balance. We've also identified Pvf1 and Upd3 as additional tumor-derived factors that activate catabolic pathways in peripheral tissues. Ongoing work focuses on discovering new tumor-secreted signals and understanding how target tissues respond. We apply transcriptomics (snRNAseq) and proteomics (proximity labeling) to uncover mechanisms driving cachexia and systemic energy imbalance in response to tumors.

Drosophila Gut and Tissue Homeostasis
Our lab studies how intestinal stem cells (ISCs) maintain tissue homeostasis using the Drosophila gut as a model. We investigate how intrinsic factors and signaling pathways regulate ISC proliferation and differentiation during regeneration and stress. Through genetic screens, we’ve identified novel roles for ion channels and microRNAs, with implications for diseases like cystic fibrosis. We developed tools to visualize real-time signaling and use single-cell RNA sequencing to map gut cell types and transcriptional networks. Our goal is to understand how signaling pathways and transcription factors integrate to coordinate stem cell behavior and maintain epithelial integrity.

Tool Development

Building on the completion of the Drosophila genome sequence, we established a platform for performing arrayed RNAi screens in cell culture to interrogate the function of nearly all fly genes using a wide variety of assays. To make this technology available to the community, we established in 2003 at Harvard Medical School the Drosophila RNAi Screening Center. Using the DRSC screening platform, the functions of the ~15,000 predicted Drosophila genes can be systematically analyzed to address questions in cell signaling, cell morphology, host-pathogen interactions, ion channel function, and many other topics. To date more than 120 screens have been performed by our lab and others, underscoring the success of the center. Over the years, we have added a number of screening reagents and developed bioinformatics tools to improve the platform. We renamed the center DRSC/TRiP-Functional Genomics Resources to better represent our current capabilities. Moreover, in 2019, we were funded to function as the Drosophila Research and Screening Center-Biomedical Technology Research Resource (DRSC-BTRR), which focuses on development of new screening and other technologies. We now have available for screening: genome-wide RNAi libraries and subset RNAi libraries (Kinase/Phosphatase; Ubiquitination; Transmembrane proteins; Transcription factors; RNA-binding proteins; Autophagy-related proteins; G-protein coupled receptors; Membrane-bound organelles; and Orthologs of human proteins for which there are FDA-approved drugs); Overexpression libraries (UAS-ORFs); and reagents for both gain-of-function (UAS-miR) and loss-of-function (UAS-miR-sponges) miRNA screens. Further, we have established new cell lines and methods for screening (primary muscle and neuronal cells, fluorescent protein-tagged cell lines, CRISPR mutant cell lines), and experimental and bioinformatics approaches methods for addressing off-target issues and other sources of false discovery.
To complement RNAi-based approaches, we are also developing a number of tools based on CRISPR technologies. We are using CRISPR to mutate or engineer cell lines that can be used for screening, and have developed efficient protocols in Drosophila cells, which are particularly challenging as they are polyploid and difficult to grow following single-cell isolation. CRISPR-generated mutant cell lines, in combination with RNAi, provide a robust platform for combinatorial screening. We have also generated stable nuclease-dead Cas9 activator (dCas9a) cell lines that can be used, after transfection with gRNAs, to perform overexpression screens - thus complementing loss of function screens. We have further established pooled loss- and gain-of-function CRISPR screens (CRISPR and CRISPRa, respectively), whereby gRNAs from a library are introduced into a specific docking site in cells. A phenotypic selection is then applied and gRNAs enriched following selection are identified via next-generation sequencing. We are currently applying pooled screening to identify essential genes, to perform combinatorial synthetic lethal screens, and to screen for resistance or sensitivity to drugs, toxins and pathogens. Finally, as an alternative to RNAi for partial loss-of-function screens using a Cas system, we have shown that Cas13 is an effective tool for CRISPRi in Drosophila cells. Advantages of CRISPRi as compared with RNAi are that the level of knockdown can be more tightly controlled and the system appears to have reduced off-target effects. Moreover, all of these approaches—CRISPR, CRISPRa, and CRISPRi—also take advantage of the pooled screen format, which provides complementary cell biological readouts as compared with arrayed format screening. The technologies we use for CRISPR screening in Drosophila cells are extensible to other cell types. One point of focus for DRSC-BTRR efforts is establishment of CRISPR pooled screening in cell lines from mosquito vectors of infectious diseases.
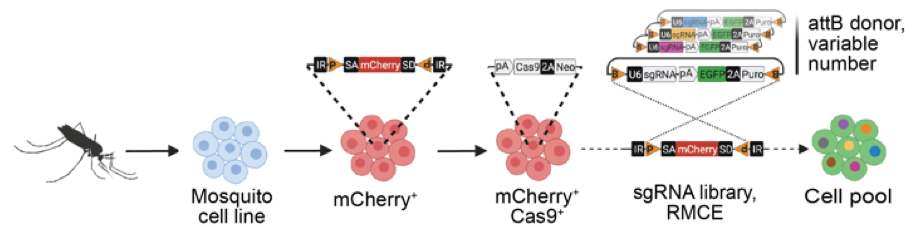
Mosquito-borne diseases, including Dengue, Zika, Chikungunya and West Nile Virus, present a worldwide public health burden. Mosquito cell lines exist that are able to become infected with viruses and are susceptible to mosquitocidal toxins, parasites, and drugs, but tools to allow deeper understanding of pathogen interactions with mosquito cells are lacking. We previously developed a recombination-mediated cassette exchange (RMCE) system that enables pooled CRISPR screening in Drosophila cells, as well as bioinformatics tools for identifying sgRNA designs for mosquitos and mosquito cell lines. We are now adapting the pooled-format CRISPR screening approach for use in cell lines from medically relevant mosquito species. With this advance, we hope to enable the mosquito community to easily perform genome-wide loss- or gain-of-function screens in cells, for example to discover entry and infection mechanisms of pathogens and drugs, as well as to aid in the functional annotation of mosquito genomes.

Proximity Labeling: Characterizing the proteome composition of organelles and subcellular regions of living cells can facilitate the understanding of cellular organization as well as protein interactome networks. Proximity labeling-based methods coupled with mass spectrometry (MS) offer a high-throughput approach for systematic analysis of spatially-restricted proteomes. Proximity labeling utilizes enzymes that generate reactive radicals to covalently tag neighboring proteins. The tagged endogenous proteins can then be isolated for further analysis by MS. To analyze protein-protein interactions or identify components that localize to discrete subcellular compartments, we developed tools based on APEX, an engineered ascorbate peroxidase derived from plants, and BioID, a mutant form of the biotin ligase BirA from E coli and demonstrated their use for in vivo studies. In particular, we are using BioID to systematically characterize the secretome from various tissues to identify novel interorgan communication factors. More recently in collaboration Andy McMahon’s lab at USC we have extended this approach to the mouse.
NanoTags: One of the key reagents to understand protein expression and function are antibodies. Antibodies allow protein visualization by immunostaining, biochemical study by immunoprecipitation and western blot, and proteomic study by IP-MS. Despite the importance of antibodies in biology study, antibodies are not available for most fly proteins. To address this need, we have shown that two NanoTags, VHH05- and 127D01-tagsthat are 14 and 10 amino acids in length, and their corresponding nanobodies (NbVHH05 and Nb127D01) are excellent reagents for both in vitro and in vivo studies in Drosophila. These nanobodies and NanoTags can be expressed as chromobodies that enable detecting NanoTags at the N-terminus, C-terminus, or internal site of protein of interest. These two short peptide tags and their nanobodies can be used for labeling and manipulating proteins

We have developed a series of bioinformatics tools that provide the research community with well-designed, user-friendly resources that impact research at all stages, from project design to data analysis and integration. These tools, which are available via DRSC/TRiP Functional Genomics Resources, include:
| DRSC/TRiP Functional Genomics Resources | ||
|---|---|---|
DIOPT DIOPT-DIST Gene2Function GLAD BioLitMine UP-TORR | RSVP SnapDragon FlyPrimerBank Find CRISPR SNP-CRISPR DGET | SNP-CRISPR DGET DRscDB COMPLEAT MIST iProteinDB |
Our most popular tools are: DIOPT, an integrative tool for ortholog predictions among major model organisms. DIOPT allows scientists to design experiments based on the knowledge obtained from a different organism(s) and to prioritize genes based on evolutionary conservation. DIOPT has been well received by the scientific community and has been integrated into FlyBase, PomBase, MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) and AGR (Alliance of Genome Resources); Gene2Function integrates annotation information for orthologs of multiple species in unified interfaces; RSVP allows scientists to mine the validation and phenotype data for in vivo RNAi and sgRNA stocks; MIST (Molecular Interaction Search Tool) for mining, visualization and analysis of network data, iProteinDB for mining protein post-translational modification (PTM) data and comparison of PTM data across species and DRscDB for mining and comparison of single-cell RNA-seq data across species.
