
")
")
")
")
")
")
")
Communication between cells, tissues and organs in Drosophila
The Perrimon lab is interested in characterizing the mechanisms by which cells, tissues and organs communicate during development and in mature animals. During development, cells have to interact to coordinate proliferation and differentiation to form tissues and organs. In mature animals, cells and organs interact to maintain homeostasis and coordinate their physiological roles. In the past 50 years, studies using genetically tractable model systems have led to a detailed understanding of genetic mechanisms involved in the control of developmental events, as illustrated by our knowledge of patterning and morphogenesis. Among the next significant questions to address are how complex phenotypes arise in the context of the whole organism and how the programs regulating these phenotypes are influenced by genetic background and environment. For example, little is understood about how the simultaneous growth and differentiation of different tissues and organs are coordinated and how the development of different cell types and tissues is integrated within an organ. Additionally, many of the mechanisms by which growth factor–triggered signaling events intersect with cell metabolism, which is regulated by nutrients, hormones and stress, remain to be identified. Acquiring answers to these fascinating questions requires a deep understanding of the mechanisms by which cells integrate signals received from their inner selves, neighboring cells, other organs and the environment. We use Drosophila as a model system to characterize the mechanisms underlying communication between cells, tissues, and organs. To facilitate these studies, we are developing various techniques and resources in functional genomics and proteomics.
Our major contributions to tool development are the Gal4-UAS system, FLP-FRT germline clone technique, in vitro and in vivo genome-wide RNAi and CRISPR screening platforms and resources, and in vivo proximity labeling methods. Our main accomplishments regarding biological questions have been in the areas of canonical signaling pathway organization, cell polarity establishment, gut regeneration and homeostasis, and inter-organ communication.
Main Research Areas (click to expand headings below for more information)
Tool development: Cell based RNAi and CRISPR
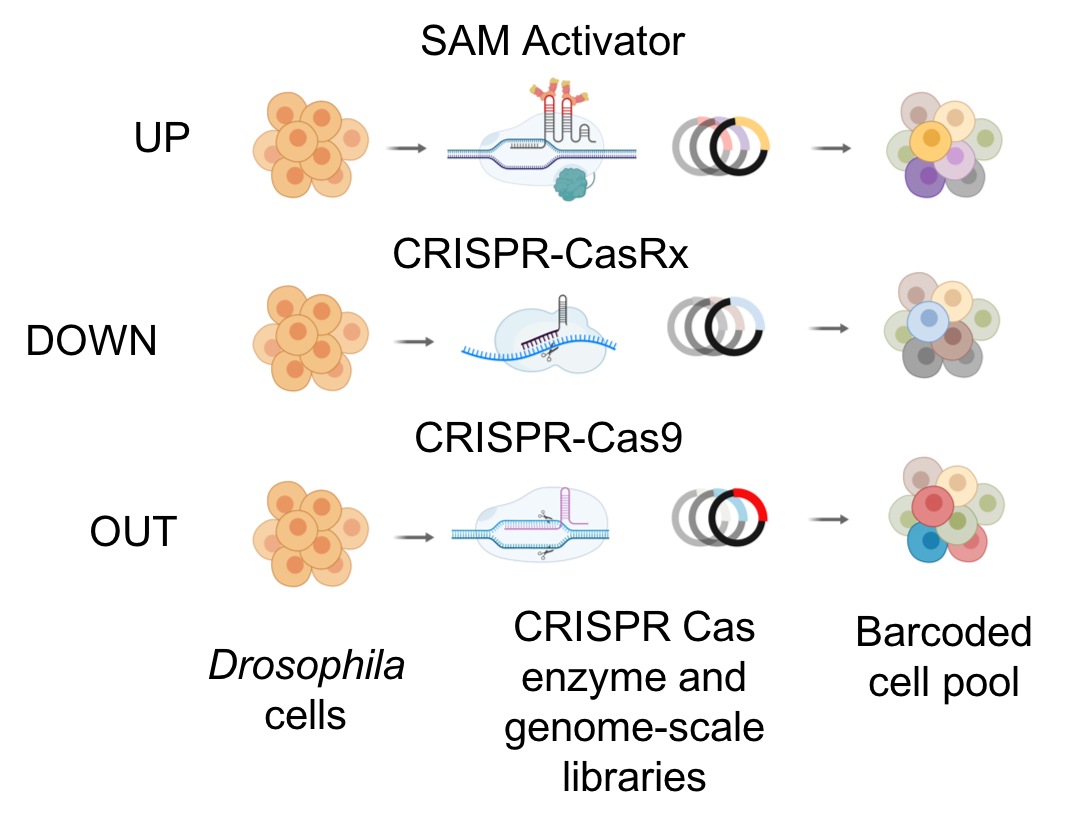 Building on the completion of the Drosophila genome sequence, we established a platform for performing arrayed RNAi screens in cell culture to interrogate the function of nearly all fly genes using a wide variety of assays. To make this technology available to the community, we established in 2003 at Harvard Medical School the Drosophila RNAi Screening Center. Using the DRSC screening platform, the functions of the ~15,000 predicted Drosophila genes can be systematically analyzed to address questions in cell signaling, cell morphology, host-pathogen interactions, ion channel function, and many other topics. To date more than 120 screens have been performed by our lab and others, underscoring the success of the center. Over the years, we have added a number of screening reagents and developed bioinformatics tools to improve the platform. We renamed the center DRSC/TRiP-Functional Genomics Resources to better represent our current capabilities. Moreover, in 2019, we were funded to function as the Drosophila Research and Screening Center-Biomedical Technology Research Resource (DRSC-BTRR), which focuses on development of new screening and other technologies. We now have available for screening: genome-wide RNAi libraries and subset RNAi libraries (Kinase/Phosphatase; Ubiquitination; Transmembrane proteins; Transcription factors; RNA-binding proteins; Autophagy-related proteins; G-protein coupled receptors; Membrane-bound organelles; and Orthologs of human proteins for which there are FDA-approved drugs); Overexpression libraries (UAS-ORFs); and reagents for both gain-of-function (UAS-miR) and loss-of-function (UAS-miR-sponges) miRNA screens. Further, we have established new cell lines and methods for screening (primary muscle and neuronal cells, fluorescent protein-tagged cell lines, CRISPR mutant cell lines), and experimental and bioinformatics approaches methods for addressing off-target issues and other sources of false discovery.
Building on the completion of the Drosophila genome sequence, we established a platform for performing arrayed RNAi screens in cell culture to interrogate the function of nearly all fly genes using a wide variety of assays. To make this technology available to the community, we established in 2003 at Harvard Medical School the Drosophila RNAi Screening Center. Using the DRSC screening platform, the functions of the ~15,000 predicted Drosophila genes can be systematically analyzed to address questions in cell signaling, cell morphology, host-pathogen interactions, ion channel function, and many other topics. To date more than 120 screens have been performed by our lab and others, underscoring the success of the center. Over the years, we have added a number of screening reagents and developed bioinformatics tools to improve the platform. We renamed the center DRSC/TRiP-Functional Genomics Resources to better represent our current capabilities. Moreover, in 2019, we were funded to function as the Drosophila Research and Screening Center-Biomedical Technology Research Resource (DRSC-BTRR), which focuses on development of new screening and other technologies. We now have available for screening: genome-wide RNAi libraries and subset RNAi libraries (Kinase/Phosphatase; Ubiquitination; Transmembrane proteins; Transcription factors; RNA-binding proteins; Autophagy-related proteins; G-protein coupled receptors; Membrane-bound organelles; and Orthologs of human proteins for which there are FDA-approved drugs); Overexpression libraries (UAS-ORFs); and reagents for both gain-of-function (UAS-miR) and loss-of-function (UAS-miR-sponges) miRNA screens. Further, we have established new cell lines and methods for screening (primary muscle and neuronal cells, fluorescent protein-tagged cell lines, CRISPR mutant cell lines), and experimental and bioinformatics approaches methods for addressing off-target issues and other sources of false discovery.
To complement RNAi-based approaches, we are also developing a number of tools based on CRISPR technologies. We are using CRISPR to mutate or engineer cell lines that can be used for screening, and have developed efficient protocols in Drosophila cells, which are particularly challenging as they are polyploid and difficult to grow following single-cell isolation. CRISPR-generated mutant cell lines, in combination with RNAi, provide a robust platform for combinatorial screening. We have also generated stable nuclease-dead Cas9 activator (dCas9a) cell lines that can be used, after transfection with gRNAs, to perform overexpression screens - thus complementing loss of function screens. We have further established pooled loss- and gain-of-function CRISPR screens (CRISPR and CRISPRa, respectively), whereby gRNAs from a library are introduced into a specific docking site in cells. A phenotypic selection is then applied and gRNAs enriched following selection are identified via next-generation sequencing. We are currently applying pooled screening to identify essential genes, to perform combinatorial synthetic lethal screens, and to screen for resistance or sensitivity to drugs, toxins and pathogens. Finally, as an alternative to RNAi for partial loss-of-function screens using a Cas system, we have shown that Cas13 is an effective tool for CRISPRi in Drosophila cells. Advantages of CRISPRi as compared with RNAi are that the level of knockdown can be more tightly controlled and the system appears to have reduced off-target effects. Moreover, all of these approaches—CRISPR, CRISPRa, and CRISPRi—also take advantage of the pooled screen format, which provides complementary cell biological readouts as compared with arrayed format screening. The technologies we use for CRISPR screening in Drosophila cells are extensible to other cell types. One point of focus for DRSC-BTRR efforts is establishment of CRISPR pooled screening in cell lines from mosquito vectors of infectious diseases.
Functional Genomics tools for Mosquitoes

Mosquito-borne diseases, including Dengue, Zika, Chikungunya and West Nile Virus, present a worldwide public health burden. Mosquito cell lines exist that are able to become infected with viruses and are susceptible to mosquitocidal toxins, parasites, and drugs, but tools to allow deeper understanding of pathogen interactions with mosquito cells are lacking. We previously developed a recombination-mediated cassette exchange (RMCE) system that enables pooled CRISPR screening in Drosophila cells, as well as bioinformatics tools for identifying sgRNA designs for mosquitos and mosquito cell lines. We are now adapting the pooled-format CRISPR screening approach for use in cell lines from medically relevant mosquito species. With this advance, we hope to enable the mosquito community to easily perform genome-wide loss- or gain-of-function screens in cells, for example to discover entry and infection mechanisms of pathogens and drugs, as well as to aid in the functional annotation of mosquito genomes.
Tool development: In vivo RNAi and CRISPR
Because results from tissue culture screens need to be followed up with in vivo validation, and given the independent value of tools for in vivo genetic studies, we have improved methods for transgenic RNAi in Drosophila. We demonstrated that shRNAs are more efficient and specific than long dsRNA when expressed as transgenes, and generated a genome scale collection of >13,000 lines covering 75% of the genome in our optimized VALIUM vectors (85% of highly conserved genes). This collection is available to the community through the Bloomington Drosophila Stock Center. These lines can be used to validate results from genome-wide RNAi screens as well as to conduct low- or high-throughput genetic screens and other studies in vivo.
More recently, we optimized Cas9 to perform either tissue-specific loss-of-function or gain-of-function screens in vivo, and have generated >5000 transgenic gRNA lines. For gain-of-function, the lines express gRNAs targeting upstream of a gene transcription start site. Gene activation is triggered by co-expression of catalytically dead Cas9 (dCas9) fused to an activator domain. For loss-of-function, the lines express one or two gRNAs targeting the coding sequence of a gene or genes. These lines can be combined with tissue-specific delivery of Cas9 to generate clones of cells (mosaics) or combined with germline expression of Cas9 to generate null mutations. In addition, these gRNA fly stock resources can be used for genome engineering of the promoter or coding regions.
We continue to evaluate and create new methods for genome engineering. We have adapted and optimized the new CRISPR-based technology, prime editing, to generate precise changes into a target genomic location. In a collaboration with Sebastian Kadener’s lab at Brandeis, we generated a new set of PspCas13b and RfxCas13d expression constructs that can be used to target RNA in cells and in vivo. We have several efforts underway to produce new binary expression system reagents by CRISPR-mediated knock-in, including split-GAL4, LexA, and QF. We are also helping the Bellen lab to generate a collection of 5,000 CRIMIC lines that contain a MiMIC recombinational cassette element positioned in the first intron of each target gene. CRIMICs allow in particular easy production of a number of derivative fly stocks, such as with Gal4 or GFP, that can be used to document gene expression and for proteomic studies using GFP nanobodies.
Tool development: Proteomics
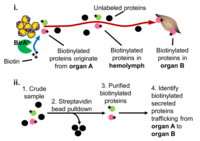 Proximity Labeling: Characterizing the proteome composition of organelles and subcellular regions of living cells can facilitate the understanding of cellular organization as well as protein interactome networks. Proximity labeling-based methods coupled with mass spectrometry (MS) offer a high-throughput approach for systematic analysis of spatially-restricted proteomes. Proximity labeling utilizes enzymes that generate reactive radicals to covalently tag neighboring proteins. The tagged endogenous proteins can then be isolated for further analysis by MS. To analyze protein-protein interactions or identify components that localize to discrete subcellular compartments, we developed tools based on APEX, an engineered ascorbate peroxidase derived from plants, and BioID, a mutant form of the biotin ligase BirA from E coli and demonstrated their use for in vivo studies. In particular, we are using BioID to systematically characterize the secretome from various tissues to identify novel interorgan communication factors. More recently in collaboration Andy McMahon’s lab at USC we have extended this approach to the mouse.
Proximity Labeling: Characterizing the proteome composition of organelles and subcellular regions of living cells can facilitate the understanding of cellular organization as well as protein interactome networks. Proximity labeling-based methods coupled with mass spectrometry (MS) offer a high-throughput approach for systematic analysis of spatially-restricted proteomes. Proximity labeling utilizes enzymes that generate reactive radicals to covalently tag neighboring proteins. The tagged endogenous proteins can then be isolated for further analysis by MS. To analyze protein-protein interactions or identify components that localize to discrete subcellular compartments, we developed tools based on APEX, an engineered ascorbate peroxidase derived from plants, and BioID, a mutant form of the biotin ligase BirA from E coli and demonstrated their use for in vivo studies. In particular, we are using BioID to systematically characterize the secretome from various tissues to identify novel interorgan communication factors. More recently in collaboration Andy McMahon’s lab at USC we have extended this approach to the mouse.
NanoTags: One of the key reagents to understand protein expression and function are antibodies. Antibodies allow protein visualization by immunostaining, biochemical study by immunoprecipitation and western blot, and proteomic study by IP-MS. Despite the importance of antibodies in biology study, antibodies are not available for most fly proteins. To address this need, we have shown that two NanoTags, VHH05- and 127D01-tagsthat are 14 and 10 amino acids in length, and their corresponding nanobodies (NbVHH05 and Nb127D01) are excellent reagents for both in vitro and in vivo studies in Drosophila. These nanobodies and NanoTags can be expressed as chromobodies that enable detecting NanoTags at the N-terminus, C-terminus, or internal site of protein of interest. These two short peptide tags and their nanobodies can be used for labeling and manipulating proteins
Bioinformatics Tools

We have developed a series of bioinformatics tools that provide the research community with well-designed, user-friendly resources that impact research at all stages, from project design to data analysis and integration. These tools, which are available via DRSC/TRiP Functional Genomics Resources, include:
|
DIOPT |
RSVP |
SNP-CRISPR DGET DRscDB COMPLEAT MIST iProteinDB |
Our most popular tools are: DIOPT, an integrative tool for ortholog predictions among major model organisms. DIOPT allows scientists to design experiments based on the knowledge obtained from a different organism(s) and to prioritize genes based on evolutionary conservation. DIOPT has been well received by the scientific community and has been integrated into FlyBase, PomBase, MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) and AGR (Alliance of Genome Resources); Gene2Function integrates annotation information for orthologs of multiple species in unified interfaces; RSVP allows scientists to mine the validation and phenotype data for in vivo RNAi and sgRNA stocks; MIST (Molecular Interaction Search Tool) for mining, visualization and analysis of network data, iProteinDB for mining protein post-translational modification (PTM) data and comparison of PTM data across species and DRscDB for mining and comparison of single-cell RNA-seq data across species.
Drosophila Gut and Tissue Homeostasis
Precise regulation of epithelial stem cells is critical to maintain tissue integrity and prevent over-proliferation and cancer. Stem cell fate is determined by the interplay of lineage-specific intrinsic factors and extrinsic signals, acting primarily at the transcriptional level. Following our identification of adult Drosophila intestinal stem cells (ISCs), we used this system to characterize genes and pathways important for gut homeostasis and in particular, to analyze crosstalk among signaling pathways.
From a number of screens (receptome-wide RNAi screen, miRNA screens, etc.), we have identified and validated novel mechanisms of how stem cells adjust their rate of proliferation/differentiation to meet the demand for tissue regeneration. For example, we identified a number of ion channels that either directly or indirectly regulate stem cell proliferation. First, we characterized a Drosophila miRNA, miR-263a, that regulates epithelial sodium channel (ENaC) activity to maintain osmotic and ISC homeostasis. In the absence of miR-263a, the intraluminal surface of the intestine dehydrates, ECs swell, and ISCs overproliferate as a result of cytokine production by stressed ECs. Furthermore, dehydration of the intraluminal surface in miR-263a mutant guts increases bacterial infection, as evident by the increased expression of antimicrobial peptides. Strikingly, these phenotypes are reminiscent of the pathophysiology of cystic fibrosis (CF), as in CF the non-functional CF transmembrane conductance regulator (CFTR) increases ENaC activity resulting in chronic dehydration of the intraluminal surface liquid in organs such as kidney, colon, lung and sweat glands. As miR-183, the human ortholog of miR-263a, also regulates ENaC, our findings suggest miR-183 might be used as a treatment for CF. Second, we showed that the calcium channel TrpA1 is expressed in ISCs and is required for their proliferation. In response to tissue damage reagents such as paraquat and bleomycin, TrpA1 mediates calcium influx, which in turn activates Ras/MAPK pathway and induces stem cell proliferation. Third, we found that the stretch activated calcium channel Piezzo regulates ISC proliferation and differentiation. Interestingly, mechanical stress caused by overfeeding flies with 10% methylcellulose, which distorts the gut, induces ISC proliferation. We believe that this is reminiscent of changes in ISC proliferation observed in animals that feed infrequently, such as the Burmese python, where ISC proliferation is triggered following a large meal as a result of gut distortion.
Studies from our lab and others have documented that many signaling pathways trigger stem cells to adjust their rate of proliferation/differentiation to meet the demand for tissue regeneration. Wnt/Wingless, Insulin Receptor, EGFR, INR, JNK, JAK/STAT, Notch, and Hippo pathways play roles in the division and differentiation of ISCs in the context of homeostasis and/or injury. The challenge is now to understand how these pathways intersect and cooperate, as it is not clear if some pathways regulate the activity of others, or if they act concomitantly. We have taken a number of approaches to address the spatial and temporal regulation of signaling pathway activities in the gut epithelium. First, we developed a new method to visualize the dynamic activation of pathway activities. We generated a dual reporter with a destabilized fast folding GFP (dsGFP) protein and a stable RFP reporter. dsGFP, because of its short half-life, captures “real-time” signaling, while stable RFP labels cells in which signaling activity has occurred. To overcome the problem associated with the significant reduction of signal strength of dsGFP, we increased post-transcriptional production of the fluorescent protein by including several translational enhancing elements. This dual reporter can be used in either fixed or live tissues to monitor spatio-temporal activity of signaling pathways at an unprecedented resolution. We are now using this tool to establish sensors of each signaling pathway involved in gut homeostasis, and will use these reagents to examine pathway crosstalk. Second, we identified 53 transcription factors, specifically expressed or enriched in the ISCs, and are characterizing how they cooperate to integrate signal inputs. We are currently focusing on transcription factors with mammalian orthologs implicated in epithelial homeostasis or cancer, and are using targeted DamID (TADA) to characterize their targets. Transcription factor Dam fusions of the major proliferative signaling pathways will be used to address questions about crosstalk and target co-regulation. Third, more recently we used single-cell RNAseq (scRNAseq) to describe all the cell types and their transcriptomes in the Drosophila gut. These studies have allowed us to better characterize what cell types are present in different gut regions and provide us with new powerful data sets with which we can better characterize the transcriptional networks that define lineage relationships among cell types, cell-cell interactions among cell types (i.e. as achieved by mapping signal pathway activities in various cell types), and the physiological roles of various cells
.
Organ wasting/Cachexia
Our studies on Drosophila gut led us to establish a model of tumor-induced organ wasting that is relevant to cachexia. Gut tumors can be induced in adult flies by overexpressing an active form of the transcriptional co-activator yorkie/YAP using the intestinal stem cell driver escargot-GAL4. These esg>yki gut tumors induce organ wasting by secreting the insulin binding protein ImpL2 into the hemolymph, which reduces insulin signaling in peripheral tissues. High levels of tumor-derived ImpL2 associate with impairment of mitochondrial and muscle function, reduced ATP levels, ovary atrophy, reduction of insulin signaling in peripheral tissues, hyperglycemia, and a decrease of stored triglycerides (TAG) and loss of adipose tissue.
In addition to ImpL2, we have also identified two additional factors produced from yki-gut tumors that contribute to tissue wasting: the Pvr receptor tyrosine kinase ligand Pvf1 and the IL6-like cytokine Unpaired 3 (Upd3). Pvf1 produced by yki-gut tumors activates MEK/ERK signaling and enhances catabolism in multiple tissues. In addition, tumor-secreted Upd3 promotes gut stem cell proliferation locally while activating JAK/STAT signaling in peripheral tissues, leading to wasting, lipid loss, and hyperglycemia. Interestingly, JAK/STAT signaling in the muscle or fat body directly triggers the expression of ImpL2, which in turn impairs local insulin signaling and its associated energy balance.
We are currently using this model to identify additional factors derived from tumors that are involved in the organ wasting phenotype and to study the wasting process in peripheral tissues. In addition, we are extending our studies to additional tumor models. Pathways that are misregulated in animals that develop organ wasting in the context of these various tumor models are identified using a combination of transcriptomics, e.g. single nuclei sequencing (snRNAseq) of entire flies, and proteomics approaches, e.g. using proximity labeling to identify all proteins secreted by the tumors. These experiments will provide a system-wide understanding of the factors originating from tumors and how they perturb systemic energy balance, and how peripheral organs in turn respond to initiate cachectic phenotypes.

Interorgan communication
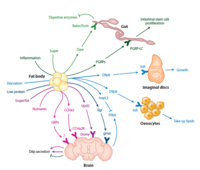 Organ-to-organ communication is critical to living systems and plays major roles in homeostasis. For example, the vertebrate CNS receives information regarding the status of peripheral metabolic processes via hormonal signaling and direct macromolecular sensing. Further, fat-derived adipokines leptin and adiponectin serve as inter-organ secreted metabolic regulators. In addition, skeletal muscles produce various myokines that influence metabolic homeostasis, lifespan, and the progression of age-related diseases and aging in non-muscle tissues. Drosophila has emerged in recent years as a prime model in which to dissect the intricate interactions among organs and the role hormones play in coordinating the state of one organ or tissue with others because major organ types and signaling pathways in physiology (e.g. leptin), neural development, and behavior are conserved. In addition and most importantly, genome-wide libraries of transgenic RNAi lines are available that allow knockdown of any Drosophila gene in an organ or tissue-specific manner. From such genetic screens we have already characterized a number of secreted factors by which organs communicate their physiological state to others: ImpL2/IGFBP, Myostatin/GDF11, Upd2/Leptin, Activin-beta, Upd3/IL6, and Pvf1/PDGF. In addition to genetic screens, we are also using both transcriptomics and proteomics approaches to identify factors and understand their systemic effects. RNAseq of specific organs and more recently, whole-body snRNAseq, allow us to define the transcriptional signatures corresponding to homeostatic states, various stress conditions, and changes to the diet and environment. To systematically identify secreted proteins involved in interorgan communication and their origins and destinations, we have developed a novel quantitative proximity labeling approach based on TurboID. Ultimately, the knowledge gained from these studies will generate testable hypotheses related to disease states such as diabetes, aging, and cancer, i.e., how biological processes observed in one tissue/organ (e.g., decreased cellular metabolism, mitochondrial dysfunction) may influence processes observed in a different tissue/organ.
Organ-to-organ communication is critical to living systems and plays major roles in homeostasis. For example, the vertebrate CNS receives information regarding the status of peripheral metabolic processes via hormonal signaling and direct macromolecular sensing. Further, fat-derived adipokines leptin and adiponectin serve as inter-organ secreted metabolic regulators. In addition, skeletal muscles produce various myokines that influence metabolic homeostasis, lifespan, and the progression of age-related diseases and aging in non-muscle tissues. Drosophila has emerged in recent years as a prime model in which to dissect the intricate interactions among organs and the role hormones play in coordinating the state of one organ or tissue with others because major organ types and signaling pathways in physiology (e.g. leptin), neural development, and behavior are conserved. In addition and most importantly, genome-wide libraries of transgenic RNAi lines are available that allow knockdown of any Drosophila gene in an organ or tissue-specific manner. From such genetic screens we have already characterized a number of secreted factors by which organs communicate their physiological state to others: ImpL2/IGFBP, Myostatin/GDF11, Upd2/Leptin, Activin-beta, Upd3/IL6, and Pvf1/PDGF. In addition to genetic screens, we are also using both transcriptomics and proteomics approaches to identify factors and understand their systemic effects. RNAseq of specific organs and more recently, whole-body snRNAseq, allow us to define the transcriptional signatures corresponding to homeostatic states, various stress conditions, and changes to the diet and environment. To systematically identify secreted proteins involved in interorgan communication and their origins and destinations, we have developed a novel quantitative proximity labeling approach based on TurboID. Ultimately, the knowledge gained from these studies will generate testable hypotheses related to disease states such as diabetes, aging, and cancer, i.e., how biological processes observed in one tissue/organ (e.g., decreased cellular metabolism, mitochondrial dysfunction) may influence processes observed in a different tissue/organ.